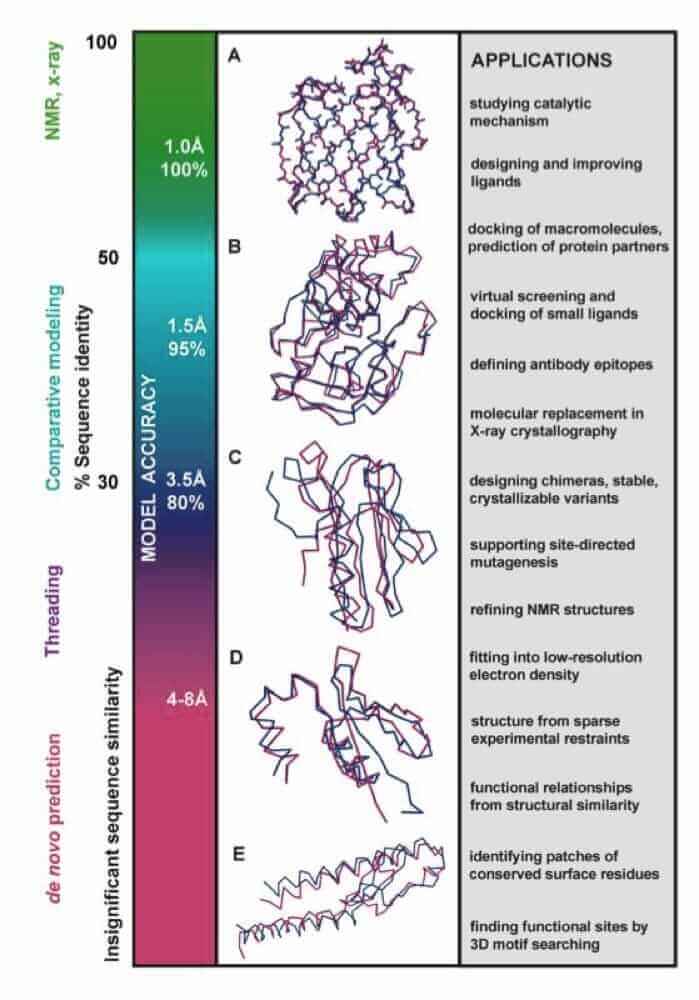
Genome sequencing projects are producing linear amino acid sequences, but full understanding of the biological role of these proteins requires knowledge of their structure and function. Although experimental structure determination methods are providing high-resolution structure information about a subset of the proteins, computational structure prediction methods will provide valuable information for the large fraction of sequences whose structures will not be determined experimentally.
The first class of protein structure prediction methods, including threading and comparative modeling, rely on detectable similarity spanning most of the modeled sequence and at least one known structure. The second class of methods, de novo or ab initio methods, predict the structure from sequence alone, without relying on similarity at the fold level between the modeled sequence and any of the known structures.
