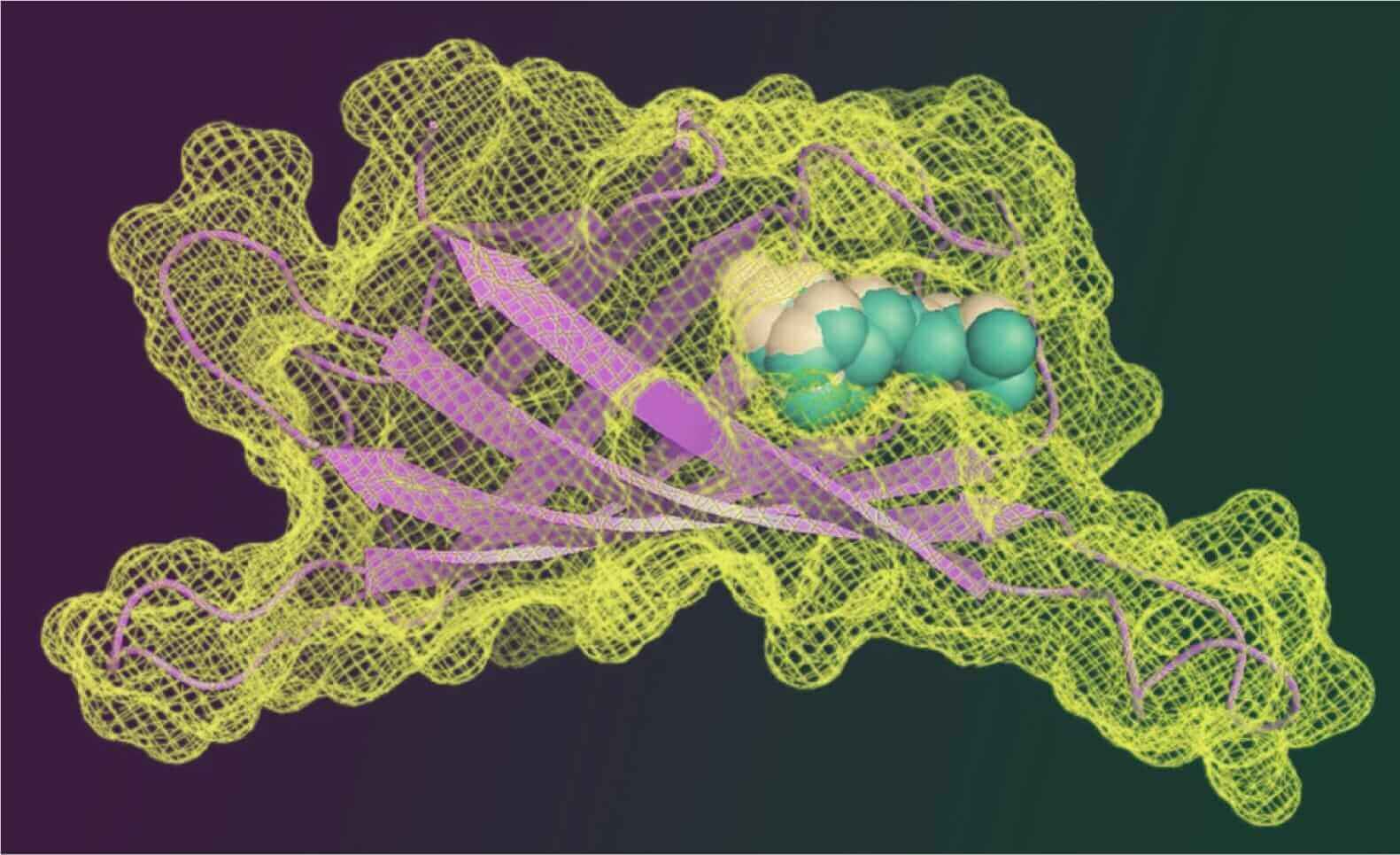
Structure-based ligand discovery begins with the atomic resolution structure of a biological receptor, and seeks to design or discover molecules that will bind to it, modulating the receptor’s function. A central technique to do so is molecular docking. In docking, libraries of about 10 7 molecules are computationally screened for those that fit the structure of the receptor (Figure 1).
For each library molecule, hundreds to thousands of orientations may be sampled in the active site of the receptor, and for every orientation hundreds of conformations; increasingly, alternate receptor conformations are also considered. Overall, about 10 13 ligand complexes may be calculated in a docking screen, each ranked using scoring functions that measure ligand- receptor complementarity.
In the physics-based methods often used at UCSF, these scoring functions measure, electrostatic (often thought of as ionic, dipole-dipole and related interactions) and van der Waals complementarity, corrected for desolvation of the ligand and the receptor. The screen results in a ranked list of candidate new ligands, almost all of which are purchasable, that may be acquired and tested in a receptor function or binding experiment.
To prosecute docking screens, investigators often turn to UCSF/QBI resources including the ZINC database of commercially available molecules, (http://zinc15.docking.org), a web-based docking program (DOCK Blaster, http://blaster.docking.org), and to comparative modeling for the structures of their receptors (Modbase, http://modbase.compbio.ucsf.edu/).

Figure 1. Molecular docking screens to discover novel ligands that can modulate a receptor.
